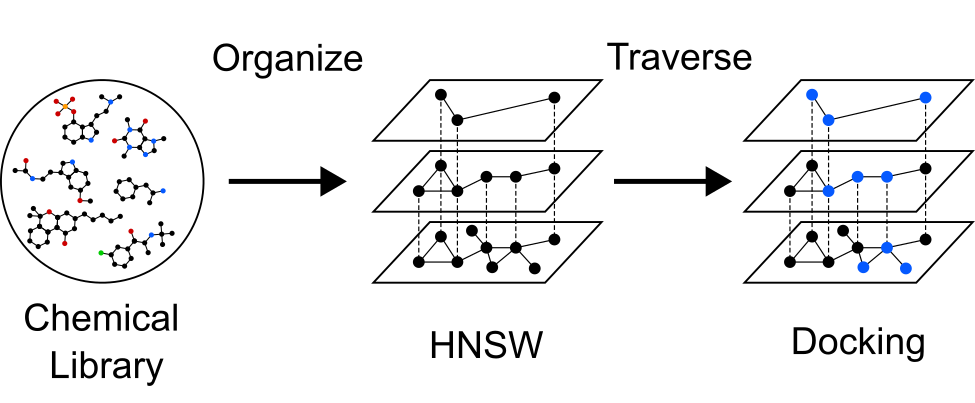
Retrieval Augmented Docking (RAD)
Molecular docking is a method to computationally predict how molecules
bind to proteins. Docking the tens to hundreds
of billions of commercially available molecules takes significant computational
resources and considerable time. We found that organizing chemical
libraries into Hierarchical Navigable Small World (HNSW) graphs
allows us to recover a majority of the best molecules while docking only a fraction of the total library.
We demonstrated this method retrospectively, screening a library of 100 million molecules
against 2 receptors with 2 different scoring functions.
For more info see the website, the
paper,
or the github repo.
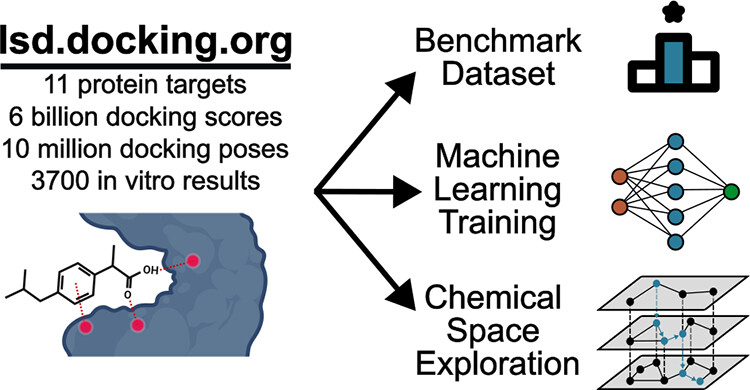
lsd.docking.org
While molecular docking campaigns of hundreds of millions to billions of molecules are performed, the results are rarely shared in full.
We developed a website providing access to poses, scores, and in vitro results for molecular docking campaigns against
11 targets, with 6.3 billion molecules docked and 3729 compounds experimentally tested.
We used the new database to train machine learning models to predict docking scores and combined these models with
my chemical space exploration method, Retrieval Augmented Docking.
For more info see the paper
or the database.
Large scale prospective evaluation of co-folding across
557 Mac1-ligand complexes and three virtual screens
While deep learning co-folding methods (Alphafold3, Boltz-2, Chai-1) can help address the challenges
of predicting ligand-bound protein complexes and ranking them, their evaluation has been hampered by data leakage and insufficiently
large test sets. We tested the ability of co-folding methods to predict the structures of 557 ligands bound to the SARS-CoV-2 macrodomain.
We also assessed whether co-folding methods could rescore molecular docking hit-lists to distinguish true ligands
from non-binders among hundreds of molecules tested against AmpC β-lactamase, and the dopamine D4 and the σ2 receptors.
For more info see the preprint.
Ribosome Profiling Data
Ribosome profiling is a method to identify ribosome pause sites within the transcriptome.
In E. Coli, the method requires an enzyme that demonstrates significant sequence
bias during RNA cleavage, hampering the ability to precisely determine the position of
ribosomes. We used 3' count assignment, pooling, and a debiasing method to more accurately
predict the location of ribosomes. We used this improved data to show that relative codon stalling rates
are not strongly correlated with relative tRNA concentrations as frequently suggested.
Preprint coming soon
This Website
One day I saw that Google was offering .phd domains and figured it was a sign
to make my own website (instead of a linkedin).
I wanted something interactive that showcased my interests in small molecule drug discovery.
I saw a cool project called 3Dmol.js which provided nice molecular visualizations and after
modifying it slightly, this is what I ended up with.
Hope you like it (and that it mostly worked while you were here) :)