My name is Brendan Hall and I'm a third year PhD student in the Biophysics
program at UCSF in the lab of Brian Shoichet (and previously Mike Keiser).
I went to Williams College where I got degrees in Physics and Math and did
research with Daniel Aalberts modeling the kinetics of translation elongation
and codon dependent ribosomal stalling.
Now at UCSF I work in structure based drug discovery and virtual screening
where I am developing methods to more efficiently traverse and score chemical space.
In my free time I enjoy playing pickup volleyball and exploring the bars
and restaurants in San Francisco.
At a high level I'm interested in how cheminformatic and machine learning
approaches can be applied to aid in the field of drug discovery with an emphasis
on virtual screening and molecular docking. As with most computational biophysics, virtual
screening comes down to two things:
1) Sampling, or how we pick molecules from the vast universe of chemical space to evaluate
2) Scoring, or how we evaluate these molecules for their desirable properties (binding affinity,
ADMET properties, etc...)
While most of my research so far has focused on how we sample molecules from chemical space (RAD)
I have a longer term interest in combining these improved sampling methods with improved scoring functions.
We still rely heavily on physics based scoring functions,
but I believe the current wealth of biophysical data will ultimately allow us to
train models that are able to more accurately and quickly predict binding affinity.
In combination with improved chemical space sampling, this will enable the
rapid and accurate screening of chemical libraries of tens of billions of molecules
and beyond.
I am currently part of the QCRG AViDD project working to apply these
methods to screen for molecules targeting SARS-Cov-2 proteins.
You can reach me by email at: brendan.hall@ucsf.edu
You can find me on twitter at: @bhall11_
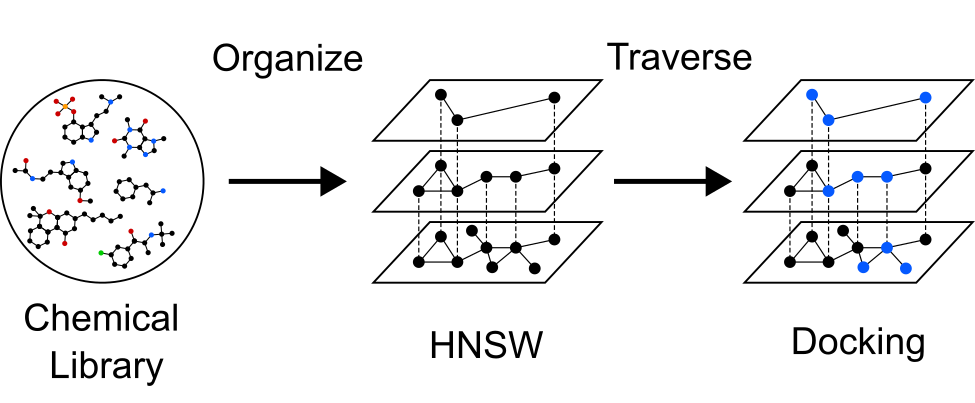
Retrieval Augmented Docking (RAD)
Molecular docking is a method that computationally predicts how well molecules
bind to proteins as a way to screen for potential drugs. Docking the tens to hundreds
of billions of commercially available molecules takes significant computational
resources and considerable time. We found that pre-organizing chemical
libraries into hierarchical navigable small world (HNSW) graphs
allows us to find a majority of the best molecules while docking only a fraction of the total library,
saving significant time, and allowing us to explore many more chemotypes. We retrospectively
demonstrated this method for screening 100 million molecules
against 2 receptors with 2 different scoring functions.
For more info see the preprint
or the github repo
Saturday Night Science
As part of a UCSF donor outreach event, I created an
interactive drug design experience to highlight how ML is being used in
drug discovery. Participants were split into groups and worked
to build physical molecular models which were then scanned via webcam to a computer where their
molecules could be docked, scored, and explored in virtual reality. All the while, they competed
against ML models which were doing high throughput virtual screening and small molecule diffusion. A central
leaderboard visualized the diffusion process, highlighted the speed at which ML methods explore
chemical space compared to humans, and showed the best scoring molecules discovered by each group and the
different ML methods.
Ribosome Profiling Data
Ribosome profiling is a method to identify the ribosome pause sites within the transcriptome.
Performing this technique in E. Coli requires an enzyme that demonstrates significant sequence
bias during RNA cleavage. This hampers our ability to precisely determine the position of the
ribosomes. We use 3' count assignment, pooling, and a debiasing method to more accurately
predict the location of ribosomes. We use this improved data to calculate relative codon stalling rates
and find that they are not strongly correlated with relative tRNA concentrations.
Preprint coming soon
This Website
One day I saw that Google was offering .phd domains and figured that was a sign
to make my own website. Rather than make a typical informational website, I wanted something
interactive that showcased my interests in small molecule drug discovery. I saw a super cool
project called 3Dmol.js which provided really slick molecular visualizations and knew that
I wanted to build my site using it. After modifying it slightly, this is what I ended up with!
Hope you like it :)